Las glomerulonefritis forman un grupo de glomerulopatías de origen inmunológico, parcialmente comprendidas y en la mayoría de los casos de tratamiento poco efectivo. Sin embargo, es de suma importancia saberlas identificar dentro de la atención del paciente con enfermedad renal. Revisamos a continuación su clasificación, patogenia y diagnóstico.
Actualízate sobre este tema en 15 minutos.
Las glomerulopatías pueden ser de origen genético, metabólico, de depósito o inmunitario. Las glomerulonefritis, en particular, son aquellas glomerulopatías de origen inmunológico en las que subyace inflamación de los glomérulos. Las glomerulonefritis se clasifican como primarias, si la enfermedad está confinada al glomérulo, o secundarias, si forman parte de una entidad sistémica. La lesión puede ser focal, cuando afecta a menos del 50% de los glomérulos, o difusa cuando abarca más del 50%. Por último, la lesión es segmentaria cuando afecta únicamente a un segmento glomerular, o global cuando afecta a todo el glomérulo.
Las principales características clínicas de la glomerulonefritis son la hematuria y en ocasiones leucocituria, a menudo con algunos eritrocitos de apariencia dismórfica, con o sin cilindros hemáticos; diversos grados de proteinuria, que va desde leve hasta el rango nefrótico; y, dependiendo de la gravedad de la enfermedad, una tasa de filtración glomerular normal o disminuida.
Inmunopatogenia de la Glomerulonefritis
La glomerulonefritis, como ya mencionamos, es ocasionada por procesos inmunológicos. Los agentes etiológicos se desconocen en su gran mayoría, con excepción de las variantes relacionadas a infección, como los estreptococos β-hemolíticos en la glomerulonefritis postestreptocócica, y el virus de la hepatitis C y B en la glomerulonefritis membranoproliferativa crioglobulinémica. Es probable que la mayoría de los factores precipitantes, como las infecciones, el cáncer y la exposición a drogas o toxinas, inicien respuestas inmunitarias similares que den lugar a glomerulonefritis a través de vías comunes.
La naturaleza de la respuesta inmune que deriva en glomerulonefritis y las personas que la desarrollan están fuertemente influenciadas por factores genéticos. La respuesta inmune nefrogénica incluye componentes tanto humorales como celulares. La respuesta humoral, regulada por linfocitos Th2, ocasiona la deposición de inmunoglobulinas y la activación del complemento en los glomérulos. La respuesta celular, regulada por linfocitos Th1 o Th17, contribuye a la infiltración de células inflamatorias mononucleares circulantes (incluidos los linfocitos y macrófagos) en los glomérulos.
Inmunidad humoral
La mayoría de las glomerulonefritis se caracterizan por la deposición de inmunoglobulinas en los glomérulos, junto con componentes del complemento, lo que sugiere que la respuesta humoral es la principal causa de lesión. Los inmunocomplejos se forman en el glomérulo, ya sea de manera activa, porque el antígeno se localice en el glomérulo, o pasiva debido al papel del glomérulo en la filtración.
Inmunidad celular
Las células mononucleares, particularmente los linfocitos y macrófagos, en ausencia de depósitos de anticuerpos, juegan un papel fundamental en la lesión glomerular en la enfermedad de cambios mínimos, la glomeruloesclerosis focal y segmentaria y algunas formas de glomerulonefritis rápidamente progresivas.
Lesión inflamatoria en la glomerulonefritis
Los dos mecanismos inmunológicos de lesión glomerular son inflamatorios y no inflamatorios. Las lesiones no inflamatorias inmunomediadas suelen afectar al podocito glomerular y están asociadas a un cambio funcional importante. La lesión inflamatoria (es decir, la glomerulonefritis) se caracteriza por hipercelularidad glomerular debido a células hematopoyéticas infiltrantes (como neutrófilos y macrófagos) y/o a células glomerulares proliferantes.
Estas células efectoras inducen otras anomalías, como la trombosis, necrosis y la formación de adherencias glomérulo-capsulares o “semilunas” que, si son extensas, pueden dar como resultado una glomerulonefritis rápidamente progresiva. Los principales mediadores y células efectoras de la respuesta inflamatoria celular en la glomerulonefritis incluyen:
- Complemento y otros mediadores humorales, como el factor inhibidor de la migración de macrófagos, quimiocinas y citocinas, que reclutan células efectoras.
- Los neutrófilos, que se adhieren al inmunocomplejo fagocitado, se activan y experimentan un estallido respiratorio con generación de especies reactivas de oxígeno, proteasas de serina catiónicas y trampas extracelulares (NET) implicadas en la necrosis glomerular.
- Macrófagos, que generan oxidantes, proteasas, factor tisular (que inicia la deposición de fibrina y la formación de medias lunas), y factor de crecimiento transformante beta (TGF-beta, que conduce a la síntesis de la matriz extracelular y al eventual desarrollo de la esclerosis).
- Células T, que liberan quimiocinas y reclutan macrófagos que posteriormente funcionan como células efectoras. Las células T helper 17 pueden ser particularmente importantes en la glomerulonefritis mediada por anti-mieloperoxidasa (MPO), presente en muchos pacientes con poliangitis microscópica y en una minoría de pacientes con granulomatosis con poliangitis.
- Plaquetas, que son prominentes en varias lesiones glomerulares, principalmente aquellas que involucran trombosis intraglomerular. Las plaquetas también liberan una serie de productos que participan y aumentan la lesión glomerular, incluidas las sustancias vasoactivas, mitogénicas y quimiotácticas.
Histopatología de la Glomerulonefritis
Las principales alteraciones que se observan en las distintas glomerulonefritis son la formación de depósitos, proliferación celular, infiltración leucocitaria, engrosamiento de la membrana basal glomerular, la hialinización y la esclerosis.
Formación de depósitos
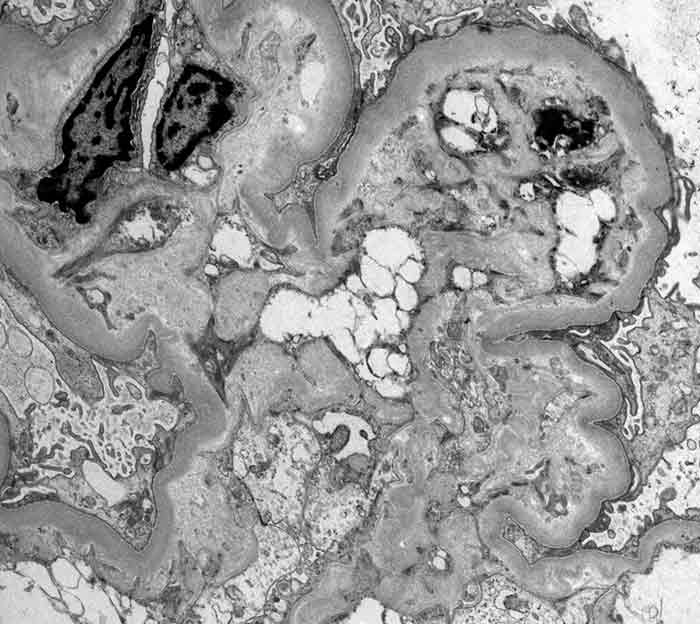
Se trata de inmunocomplejos que quedan atrapados o depositados en el glomérulo. Dependiendo de su localización se clasifican en mesangiales, subendoteliales (entre el endotelio y membrana basal), subepiteliales (entre membrana basal y epitelio visceral) e intramembranosos (dentro de la membrana basal). La localización de los inmunocomplejos se puede determinar mediante microscopía electrónica, mientras que su composición (IgG, IgA, C3, etc) y morfología (granular o linear) mediante inmunofluorescencia.
Prácticamente todas las glomerulonefritis tienen un patrón granular, mientras que los autoanticuerpos antimembrana basal glomerular conforman un patrón lineal, como en el síndrome de Goodpasture.
Proliferación Celular
La proliferación celular del glomérulo puede ocurrir en cualquiera de sus tres componentes celulares, es decir, las células endoteliales, las mesangiales y las epiteliales. Cada una de ellas responde a estímulos específicos, predominando en una glomerulonefritis determinada. Por ejemplo, en la glomerulonefritis postestreptocócica ocurre una proliferación endotelial, de ahí la clasificación en proliferativa endocapilar. De manera análoga, en la glomerulonefritis mesangial por IgA la proliferación es de las células mesangiales.
Clasificación de las Glomerulonefritis Primarias
En función de su evolución, las glomerulonefritis primarias se pueden clasificar de la siguiente manera:
Acorde al tipo de agresión inmunológica, se pueden diferenciar 3 grupos de glomerulonefritis primarias:
- No es posible detectar ningún depósito inmunológico en el glomérulo.
- GN extracapilar tipo III (pauciinmune).
- GN de cambios mínimos.
- Glomeruloesclerosis focal y segmentaria.
- Formación intrarrenal de complejos antígeno-anticuerpo.
- GN extracapilar tipo I y enfermedad de Goodpasture.
- GN membranosa.
- Atrapamiento glomerular de inmunocomplejos circulantes Ag-Ac.
- GN endocapilar (GN Aguda Postinfecciosa).
- GN extracapilar tipo II.
- GN mesangial por IgA.
- GN membranoproliferativa tipo I
- GN mesangial por IgG y/o C3.
- GN con depósitos mesangiales aislados de IgM.
Glomerulonefritis Aguda Postinfecciosa
Esta glomerulonefritis endocapilar aguda se presenta posterior a la infección por determinadas bacterias. La más relevante es la ocasionada por el estreptococo beta-hemolítico del grupo A, en cuya estructura existen antígenos que pueden inducir la formación de inmunocomplejos. Al depositarse en el glomérulo, estos complejos producen lesión o la de moléculas como el receptor de plasmina asociado a nefritis (NAPIr). Estos inmunocomplejos actúan como superantígenos induciendo la proliferación y activación de células T y liberación de mediadores inflamatorios.
Cabe resaltar que esta glomerulonefritis puede presentarse como una complicación, tanto de la faringitis estreptocócica como del impétigo, mientras que la fiebre reumática se presenta únicamente después de una faringitis por esta bacteria. La glomerulonefritis postestreptocócica se caracteriza por un periodo de latencia hasta el inicio de la presentación clínica. La cual se caracteriza por hematuria, proteinuria, oliguria e hipertensión arterial. Momento para el cual ya se han formado suficientes inmunocomplejos capaces de lesionar al glomérulo. La activación del complemento en esta glomerulonefritis es por la vía alterna, con disminución de C3.
Anatomía Patológica
No está indicada la realización de biopsia renal en la glomerulonefritis postestreptocócica, a menos que se sospeche otra etiología o la presencia de proliferación extracapilar, por insuficiencia renal rápidamente progresiva. Las indicaciones de la biopsia renal son:
- Glomerulonefritis con descenso del complemento y síndrome nefrítico, como LES o crioglobulinemia, sin asociación infecciosa.
- Complemento que persiste bajo por más de 8 semanas.
- Microhematuria por más de 6 meses.
- Oliguria por más de 3 semanas.
En la microscopía óptica se observa un glomérulo con diferentes grados de edema, hipercelularidad, proliferación de células endoteliales y mesangiales e infiltración neutrofílica (pus) y linfocitaria en el intersticio. En la inmunofluorescencia se observa depósito de C3, IgG e IgM con patrón en “cielo estrellado” distribuidos a lo largo del mesangio y asas capilares. En la microscopía electrónica se observan depósitos electrodensos en la zona subepitelial en forma de jorobas o “humps”, particulares de la glomerulonefritis postinfecciosa.
Presentación Clínica
El paciente se presenta típicamente con un síndrome nefrítico de inicio súbito y antecedente de infección, con un periodo de latencia de dos a tres semanas en la faringitis estreptocócica y de cuatro a seis semanas en el impétigo. Esto es de utilidad para el diagnóstico diferencial con la glomerulonefritis mesangial, en la que la hematuria concurre con la infección. Menos del 1% desarrolla proliferación extracapilar con insuficiencia renal rápidamente progresiva.
La disminución característica de C3 se normaliza a las 6 a 8 semanas, mientras que las alteraciones del sedimento, como la hematuria, se normalizan a los seis meses. Para el diagnóstico se requieren dos de los siguientes criterios:
- Cultivo positivo de una cepa nefritogénica de estreptococo beta-hemolítico del grupo A (cepas 1, 2, 4, 12, 18, 25, 49, 55, 57 y 60), de muestra tomada de un foco faríngeo o cutáneo.
- Anticuerpos antiestreptolisina (ASLO) elevados.
- Descenso transitorio de C3 con normalización a las 8 semanas.
Tratamiento y Pronóstico
No existe un tratamiento específico para la glomerulonefritis postinfecciosa, con excepción de las medidas de soporte del síndrome nefrítico y antibióticos. El paciente tiene una evolución favorable y recuperación completa en el 95% de los casos, habiendo mayor repercusión en adultos que en niños.
La glomerulonefritis extracapilar se caracteriza por la proliferación de la célula epitelial hacia el espacio extracapilar, con formación de semilunas en el glomérulo. Esta glomerulonefritis ocurre cuando la membrana basal glomerular se ve lesionada y queda gravemente dañada, lo que genera el paso de fibrinógeno hacia el interior de la cápsula de Bowman. El fibrinógeno, a su vez, sirve probablemente como estímulo mitógeno para las células epiteliales, las cuales proliferan y forman las semilunas en el espacio de Bowman.
Las semilunas pueden ser epiteliales o fibrosas, afectando al glomérulo de manera segmentaria, y cuyo crecimiento puede comprimir al ovillo capilar. La glomerulonefritis extracapilar se clasifica en tres variantes, las cuales se diferencian por el estímulo proliferativo y los marcadores periféricos, es decir, presencia de autoanticuerpos, inmunocomplejos, descenso de complemento, entre otros.
Glomerulonefritis Extracapilar Tipo I
Se caracteriza por la presencia de anticuerpos antimembrana basal, formados por la alteración del colágeno tipo IV alveolar por hidrocarburos, virus u otros tóxicos pulmonares. Ello ocasiona la reacción cruzada contra el colágeno tipo IV de la membrana basal glomerular, lesión de ésta última y salida de fibrinógeno al espacio subepitelial con formación de semilunas. Puede presentarse de manera aislada o asociada a hemorragia alveolar, en cuyo último caso se denomina enfermedad de Goodpasture.
En la microscopía óptica se pueden observar las semilunas, mientras que en la inmunofluorescencia es posible detectar depósitos lineales de IgG a nivel de la membrana basal glomerular. Es posible la detección sérica de los anticuerpos antimembrana basal glomerular y el complemento es normal.
Glomerulonefritis Extracapilar Tipo II y III
La tipo II puede ocurrir como complicación de una glomerulonefritis primaria (endocapilar o mesangiocapilar) o ser secundaria a una enfermedad sistémica, como lupus eritematoso sistémico o crioglobulinemia mixta esencial. En la inmunofluorescencia se pueden observar depósitos de IgM y C3 en asas capilares y mesangio. Ocurre un descenso del complemento y se pueden detectar inmunocomplejos circulantes.
Glomerulonefritis Extracapilar Tipo III
La tipo III es secundaria a una vasculitis sistémica tipo ANCA, mieloperoxidasa o preoteinasa 3 (PR3). La inmunofluorescencia es negativa, detectando únicamente fibrinógeno. Los marcadores periféricos de utilidad son los anticuerpos anticitoplasma de neutrófilos (p-ANCA, c-ANCA). El complemento es normal.
Así se presenta tu paciente
La glomerulonefritis extracapilar se presenta con mayor frecuencia en hombres, con amplia distribución de la edad. Los pacientes presentan insuficiencia renal rápidamente progresiva, síndrome nefrítico y constitucional. Cabe resaltar que cualquiera de las tres variantes de la glomerulonefritis extracapilar puede asociar hemorragia alveolar.
Tratamiento y Pronóstico
El pronóstico es desfavorable, en particular ante la presencia de semilunas fibrosas o afectación glomerular extensa. El tratamiento debe ser inmediato y agresivo, mediante rituximab como de elección. La plasmaféresis está reservada para los casos graves, en especial si asocian hemorragia pulmonar. El 50% de los pacientes terminan recibiendo diálisis a los 6 meses del inicio de la enfermedad.
Enfermedad de Cambios Mínimos
La enfermedad de cambios mínimos es de etiología aún desconocida y se presenta principalmente en niños; además, es causa del 70 a 90% de los casos de síndrome nefrótico en menores de 10 años, con mayor incidencia en niños entre los 2 y los 6 años.
Anatomía Patológica
En la microscopía óptica el glomérulo se observa normal, mientras que los túbulos contienen vacuolas de lípidos y proteínas al interior de las células epiteliales, lo que se conoce como nefrosis lipoidea. Los vasos y el intersticio no presentan alteraciones. En la inmunofluorescencia hay ausencia de depósitos y en la microscopía electrónica existe borramiento de los pedicelos debido a su fusión con las células del epitelio visceral. Cabe resaltar que esta lesión es característica de la enfermedad de cambios mínimos, sin embargo, puede presentarse en otras entidades que cursan con proteinuria importante.
Así se presenta tu paciente
Como ya mencionamos, los pacientes con enfermedad de cambios mínimos cursan con síndrome nefrótico, así como alteraciones del sedimento. La proteinuria consiste principalmente de albúmina y en el 20 a 30% de los casos se presenta microhematuria. El complemento se encuentra normal.
Tratamiento y Pronóstico
El tratamiento consiste en corticoides, siendo también de utilidad en las recidivas. Ante la falta de respuesta a dicho tratamiento se debe considerar un diagnóstico alternativo, como la glomerulosclerosis focal y segmentaria, y estará indicada la biopsia renal con inclusión de nefronas yuxtaglomerulares.
Glomeruloesclerosis Focal
La glomeruloesclerosis focal puede ser primaria o secundaria. La primaria, a su vez, puede ser idiopática o de origen genético. La lesión característica de la glomeruloesclerosis primaria, como su nombre lo dice, es la esclerosis segmentaria del glomérulo, con afectación de < 50% de los glomérulos (focal) e inicio en la zona yuxtamedular. En los casos de origen genético, el principal defecto está en el gen NPHS2, el cual codifica para la podocina. Proteína cuya función es indispensable para el correcto funcionamiento de la barrera de permeabilidad podocitaria.
La glomeruloesclerosis focal y segmentaria es la glomerulonefritis que más rápido recidiva posterior al trasplante renal, lo que sugiere factores extrarrenales implicados en la etiopatogenia de la lesión idiopática.
Glomeruloesclerosis Focal Secundaria
La glomeruloesclerosis focal secundaria puede asociar hiperfiltración, con o sin disminución de la masa renal funcionante, o toxicidad directa para el podocito. En ambos casos ocurre la pérdida de este elemento glomerular, denudación de la membrana basal, su posterior fusión con la cápsula de Bowman y, por último, esclerosis glomerular. Esto se manifiesta por proteinuria en rango nefrótico. La albúmina juega un papel primordial en el proceso de la glomeruloesclerosis, dado que esta proteína se considera tóxica para la célula epitelial y, por tanto, sirve como marcador de daño renal en la clínica.
De igual manera, contribuyen a este proceso la angiotensina II, las citocinas proinflamatorias y los factores de crecimiento.
| Hiperfiltración con disminución de masa renal funcionante | Hiperfiltración con masa renal conservada | Toxicidad directa al podocito |
Nefrectomía
Hipoplasia, agenesia o displasia renal
ERC de cualquier etiología | Anemia de células falciformes
Diabetes mellitus
Obesidad
Síndrome de Apnea Obstructiva del Sueño | VIH, P. falciparum, Schistosoma haematobium, Heroína |
Anatomía Patológica
En la microscopía óptica es posible observar engrosamiento del asa capilar, con depósito de material hialino en un segmento del ovillo glomerular. Dado que los glomérulos yuxtamedulares son los de mayor filtración, son también los que se ven más afectados. Puede haber atrofia tubular y fibrosis intersticial progresiva. Se observa esclerosis con formación de sinequias entre el epitelio visceral y el parietal en la cápsula de Bowman. Acorde a Columbus la glomeruloesclerosis segmentaria y focal se clasifica en:
- Clásica o no especificada
- Perihiliar
- Lesión del dominio tubular (Tip)
- Celular
- Colapsante
La inmunofluorescencia es generalmente negativa y en la microscopía electrónica se observa colapso focal de las membranas basales y denudación de la superficie epitelial. Existe borramiento de las proyecciones epiteliales de los podocitos.
Así se presenta tu paciente
La glomeruloesclerosis focal y segmentaria, se presenta entre los 16 y 30 años de edad y cursa con síndrome nefrótico en el 60% de los casos, además de microhematuria e hipertensión arterial. La glomeruloesclerosis focal secundaria a diabetes mellitus u obesidad no presenta hipoalbuminemia ni edema, a pesar de proteinuria importante. Por tanto, si el paciente presenta hipoalbuminemia y más del 80% de daño podocitario, se debe sospechar de glomeruloesclerosis focal y segmentaria primaria.
Tratamiento y Pronóstico
Tanto en la enfermedad primaria como en la secundaria el tratamiento debe incluir IECAs o ARA II por al menos 6 meses para disminuir la proteinuria y el tratamiento del síndrome nefrótico asociado. En la primaria, se indican corticosteroides durante 8 a 12 semanas, con remisión de la proteinuria en el 25% de los casos. Como ya mencionamos, la glomeruloesclerosis es la glomerulonefritis que más rápido recidiva posterior al trasplante renal. La evolución es lenta y progresiva y lleva a enfermedad renal terminal.
Glomerulonefritis Membranosa
La glomerulonefritis membranosa es la principal causa de síndrome nefrótico en adultos y se caracteriza por la formación in situ de complejos antígeno-anticuerpo, debido a un antígeno en la membrana basal glomerular. Se clasifica en primaria y secundaria. La primaria es ocasionada por la formación de autoanticuerpos contra la fosfolipasa A2 (PLA2R) en la vertiente subepitelial del podocito. La glomerulonefritis membranosa secundaria es ocasionada, ya sea por un componente de la membrana basal glomerular modificado por un fármaco o virus, o un antígeno extrarrenal proveniente de neoplasias o infecciones y situado en el glomérulo.
En ambas variantes de la glomerulonefritis membranosa, el inmunocomplejo se forma en la vertiente subepitelial de la membrana basal glomerular. La formación in situ ocasiona la proliferación de la membrana basal por células epiteliales (spikes), con alteración de la permeabilidad de la barrera y proteinuria en rango nefrótico.
Anatomía Patológica
La membrana basal proliferante termina por cubrir los inmunocomplejos. En las formas secundarias existe hipercelularidad mesangial o depósitos mesangiales o subendoteliales. En la microscopía óptica se observa dicho engrosamiento de la membrana basal, con afectación global glomerular y material PAS positivo. En la inmunofluorescencia se pueden identificar depósitos granulares de IgG y C3 en la vertiente subepitelial de la membrana basal glomerular. La identificación de múltiples tipos de inmunoglobulinas (IgG, IgM, IgA) es sugerente de glomerulonefritis membranosa secundaria.
La microscopía electrónica permite clasificar la enfermedad en cuatro fases, siendo la I y II las únicas reversibles. En la fase I los depósitos subepiteliales son pequeños y corresponden a los inmunocomplejos formados in situ. En la fase II se observan los spikes de la membrana proliferante que comienzan a rodear a los inmunocomplejos, mientras que en la III ya están prácticamente englobados. En la fase IV el ensanchamiento de la membrana basal glomerular es evidente, con áreas de rarefacción y pérdida de densidad de los depósitos.
Así se presenta tu paciente
La glomerulonefritis membranosa tiene su máxima incidencia entre los 30 y 50 años, con predominio en varones. La mayoría de los casos se presentan con síndrome nefrótico de comienzo insidioso y en el 30% de los casos asocia insuficiencia renal. En todo adulto con síndrome nefrótico se debe realizar biopsia renal, debido a otras entidades que pudieran provocarlo. Además, se debe evaluar la presencia de neoplasias o infecciones subyacentes. Los niveles del complemento son normales.
Tratamiento y Pronóstico
En adultos, la glomerulonefritis membranosa tiene una tasa de remisión espontánea del 20 a 30%. El tratamiento consiste en corticoides e inmunosupresores en casos de alto riesgo, es decir, con proteinuria e hipoalbuminemia graves, edema importante y/o insuficiencia renal. La enfermedad puede recidivar en el trasplante renal. Se debe brindar tratamiento del síndrome nefrótico a todos los pacientes. En niños el pronóstico es mucho mejor, con tasas de supervivencia a los cinco años mayores al 90% y remisión espontánea completa dentro de los primeros 5 años.
Glomerulonefritis Mesangial por IgA
La glomerulonefritis mesangial por IgA o enfermedad de Berger es la glomerulonefritis más frecuente a nivel mundial y predomina en varones. Tiene asociación familiar, así como a HLA-BW35. Esta glomerulonefritis se caracteriza por un aumento en la síntesis de IgA secundaria a la estimulación antigénica, así como depósito de IgA y complemento a nivel glomerular, con proliferación mesangial y activación de mediadores inflamatorios. Ocurre alteración del aclaramiento hepático de dichos anticuerpos en pacientes con producción de una IgA anormal o con cirrosis.
Anatomía Patológica
En la microscopía óptica se observa proliferación mesangial focal o difusa, mientras que en la inmunofluorescencia aparecen depósitos de IgA en mesangio, junto con C3 y properdina. En la microscopía electrónica se observan depósitos electrodensos mesangiales.
Presentación Clínica
Se presenta principalmente en hombres entre la segunda y tercera década de vida. El paciente refiere característicamente hematuria (macroscópica) recidivante. La hematuria generalmente coincide con una infección de vías respiratorias (desencadenante antigénico) o el ejercicio en las primeras 24 hrs., lo que ayuda a distinguirla de la glomerulonefritis aguda postinfecciosa. La presentación puede ser subclínica, con proteinuria y/o hematuria, así como síndrome nefrítico o nefrótico.
En niños, además del depósito a nivel glomerular, puede haber afectación por inmunocomplejos a nivel articular, gastrointestinal y púrpura, lo que se denomina síndrome o púrpura de Henoch-Schönlein. Por otro lado, cabe resaltar que solo el 50% de los pacientes presentará elevación de la IgA plasmática, mientras que los niveles del complemento se mantienen normales.
Tratamiento y Pronóstico
Se recomienda el control estricto de la presión arterial con meta de < 125/75 mmHg en pacientes con proteinuria, estando indicados los IECAs o ARA II. No se recomienda el uso combinado de estos fármacos ya que pueden provocar lesión renal aguda, siendo la monoterapia igual de efectiva. La budesonida modificada se activa localmente en el tejido linfoide del íleo distal y el ciego, con disminución de la producción de IgA. Este fármaco ha demostrado resultados positivos en pacientes con proteinuria > 0.5-0.75gr./24 horas y TFG >45 ml/min con una disminución del deterioro de la función renal. Se consideran factores de mal pronóstico los siguientes:
- Proteinuria mayor a 0.5 g/día.
- Hipertensión arterial.
- Ausencia de brotes de hematuria macroscópica.
- Insuficiencia renal en el momento del diagnóstico.
- Diagnóstico en edad adulta y varones.
- Síndrome metabólico asociado.
- Polimorfismo del gen de la enzima convertidora de angiotensina.
Para profundizar acerca de esta glomerulonefritis te recomendamos esta otra revisión.
Glomerulonefritis Mesangiocapilar
Se conoce también como glomerulonefritis membranoproliferativa y cursa con importante proliferación mesangial difusa y engrosamiento capilar; así como imágenes de doble contorno de la membrana basal glomerular debido a la interposición del citoplasma de la célula mesangial. Se diferencian la tipo I y II (actualmente enfermedad por depósitos densos). Además, se clasifican en primarias o idiopáticas y las secundarias.
La glomerulonefritis mesangiocapilar se asocia a infecciones, como la del VHC con crioglobulinemia, VHB, VIH, endocarditis infecciosa, abscesos, shunt AV infectado y la malaria. Dentro de las enfermedades autoinmunes asociadas destacan el lupus eritematoso sistémico, síndrome de Sjögren, enfermedad mixta del tejido conectivo y la crioglobulinemia.
La glomerulonefritis mesangiocapilar tipo I se desarrolla en presencia de un antígeno crónico, con formación de inmunocomplejos y gran activación del complemento. Ello ocasiona daño renal por acumulación de gran cantidad de inmunocomplejos, tanto en el mesangio como subendotelial, así como por activación del complemento por la vía clásica, con descenso de C3, C4 y C1q. En la enfermedad por depósitos densos (antes tipo II), por el contrario, el daño ocurre por una activación anómala de la vía alterna del complemento, con descenso únicamente de C3 y presencia del C3Nf (C3 nephritic factor).
Anatomía Patológica
En la glomerulonefritis mesangiocapilar tipo I se observa en la microscopía óptica un aumento de la matriz mesangial con expansión de la circunferencia entre la membrana basal glomerular y la célula endotelial. Ello genera una imagen de doble contorno o en “vías de tren” de la membrana basal. En la inmunofluorescencia se observan depósitos granulares de IgG, IgM y C3 en el mesangio y a nivel subendotelial. En la enfermedad por depósitos densos, por el contrario, se observan depósitos de C3 intramembranosos y sin depósitos de inmunoglobulinas.
Presentación Clínica
Las formas primarias de esta glomerulonefritis ocurren con mayor frecuencia en pacientes entre los 5 y 30 años de edad. La mayoría de los casos se presenta con síndrome nefrótico e hipocomplementemia. El síndrome nefrítico es más frecuente en la glomerulonefritis mesangiocapilar asociada a otras enfermedades autoinmunes. La enfermedad por depósitos densos se asocia a lipodistrofia parcial, anemia hemolítica y retinopatía, con drusas, degeneración macular y amaurosis. La hipertensión arterial grave es un hallazgo común y los pacientes cursan con deterioro progresivo de la función renal.
Tratamiento y Pronóstico
No existe un tratamiento curativo para la glomerulonefritis membranoproliferativa. Los corticoides pueden ser de utilidad en algunos casos, especialmente en niños. En formas agresivas, en especial aquellos con proliferación extracapilar, los corticoides asociados a ciclofosfamida o micofenolato mofetil, así como la plasmaféresis, pueden ser de utilidad. En la enfermedad por depósitos densos el tratamiento depende de la presencia del C3Nf, en cuyo caso estará indicada la plasmaféresis, así como de la deficiencia o ausencia del factor H.
En caso de deficiencia genética del factor H se usa plasmaféresis; por el contrario, si se trata de un autoanticuerpo contra dicho factor, se usa plasmaféresis más rituximab. El eculizumab es un inhibidor de C5, el cual está indicado en las glomerulonefritis asociadas a alteración de la vía alterna del complemento. La enfermedad por depósitos densos tiene muy mal pronóstico y recidiva invariablemente posterior al trasplante renal.
Referencias Bibliográficas
Floege J, Barbour SJ, Cattran DC, et al. Management and treatment of glomerular diseases (part 1): conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2019;95(2):268-280.
KDIGO Clinical Practice Guideline for Glomerulonephritis. Work Group Membership. Kidney Int Suppl (2011). 2012;2(2):141.
Nishi S, Ubara Y, Utsunomiya Y, et al. Evidence-based clinical practice guidelines for nephrotic syndrome 2014. Clin Exp Nephrol. 2016;20(3):342-70.
Lombel RM, Gipson DS, Hodson EM. Treatment of steroid-sensitive nephrotic syndrome: new guidelines from KDIGO. Pediatr Nephrol. 2013;28(3):415-26.
Hahn BH, Mcmahon MA, Wilkinson A, et al. American College of Rheumatology guidelines for screening, treatment, and management of lupus nephritis. Arthritis Care Res (Hoboken). 2012;64(6):797-808.
El artículo Glomerulonefritis: Revisión de su Clasificación, Patogenia y Diagnóstico. apareció primero en Sapiens Medicus.